

「若20世紀是藥物治療時代,那21世紀將會是細胞治療的時代」,近年來刮起一陣再生醫療風暴,從早期的化學藥物,發展至現今使用細胞回輸人體的新興治療技術。
與傳統製藥有別,細胞治療目前遇到的困境與挑戰
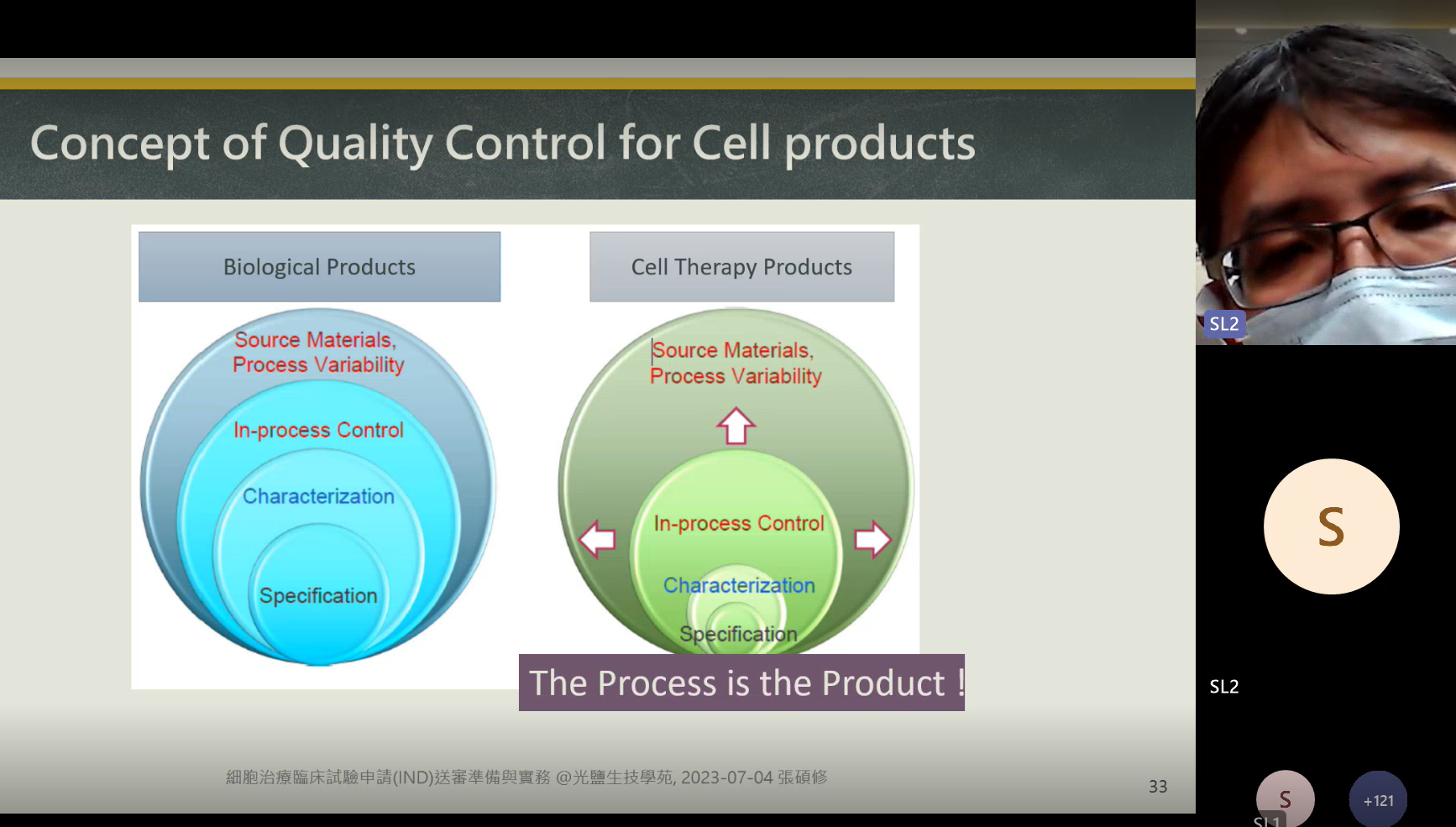
從小分子藥(化學藥)演進到大分子藥(蛋白藥)已成為全球製藥產業必然的發展過程。細胞治療在基於蛋白藥的基礎上更進一步,無論是在原料、製程、產品面都有截然不同的差異。對於一個新興技術的誕生,伴隨著的除了要證明其療效及安全性,還要符合倫理考量,而這一切也都要在法規框架內被嚴格監控。
舉例3點細胞治療產品與傳統藥物的差異,不難看出還有一大段路要走:
- 原料層面:細胞治療的關鍵原物料品質可能因個體差異而有很大的變異性,使得品質控制變得異常困難。
- 製程層面:因細胞製程手工操作比例高,存在一定的污染風險,所以對於廠房設備要求高。
- 產品層面:相對於傳統藥物而言,細胞治療產品製造批量小,但批次間變異性大,較難掌握其穩定性。

大部分廠商在申請細胞治療IND時,常因CMC文件缺失而被要求補件!
細胞治療產品在正式進入臨床試驗階段前,需要通過以下2大送審手續:
- 向衛生福利部食品藥物管理署(TFDA)提交IND(Investigational New Drug)申請
- 向醫院(site)提交IRB(Institutional Review Board)申請
在提交IND臨床試驗申請時,除了TFDA會安排查廠外,廠商還需要準備技術性文件提供審查,資料包含:CMC、非臨床試驗(動物實驗)及臨床試驗相關資料。張處長以其曾擔任過CDE審查員的經驗表示:第一次送審IND的廠商幾乎都會在CMC(Chemistry, Manufacturing and Controls)階段卡關。
藉由此次的線上分享,張處長詳細說明從原料到最終成品(含容器)的管控資訊,值得注意的是:CMC資料不只局限於原料取得及製備過程的管控,也需涵蓋到成品出貨後的管控,例如運送及醫療場所暫存條件等資料。

因再生製劑原料特性,針對生物性原料/輔料的管控應更加嚴謹!
考量到再生製劑產品無法進行最終滅菌的程序,製程中也無法執行病毒清除、去活化等步驟,所以對於生物性原料的管控也相對嚴謹,有興趣者可參考CDE於111年11月更新的《製程中使用生物性原料之研發策略指導原則(第二版)》。
為使學員能快速了解當中的規定,張處長從中整理了幾點重要的注意事項並一一說明:
- 輔料風險分級與範例
- 不得以需使用第四級物料時之作法
- 生物性原料的管控
- 人類血液來源之原料的管控
- 動物性來源之原料的管控
- 以人源或動物性來源物質所製成之原料的管控
- 重組DNA技術製造生產之蛋白質原料或病毒載體
- 再生醫療產品原料/輔料相關法規



